论文总字数:14916字
目 录
第一章. 绪论 3
1.1 二维材料介绍 3
1.2 蓝磷介绍 3
1.3 黑磷介绍 3
1.4 本文的工作 4
第二章. 少数层蓝磷的电子能带结构和态密度 5
2.1 计算方法 5
2.1.1 第一性原理计算方法 5
2.1.2 密度泛函理论 5
2.2 单层蓝磷电子能带结构和态密度 7
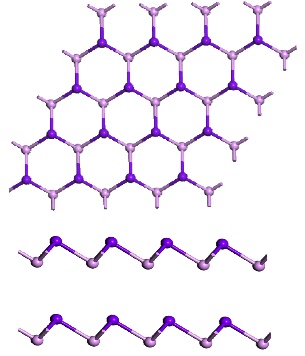
2.2.1 单层蓝磷的晶格结构 7
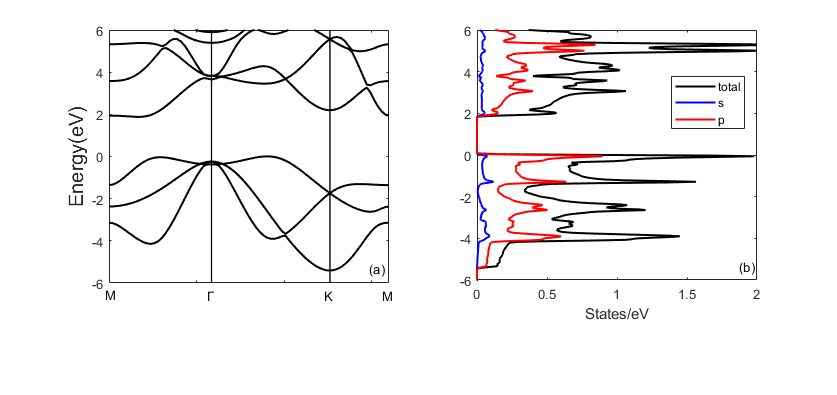
2.2.2 计算结果与讨论 8
2.3 堆叠结构对双层蓝磷的电子能带结构和态密度的影响 8
2.3.1 双层蓝磷的晶格结构 9
2.3.2 AA堆叠双层蓝磷的电子能带结构和态密度计算结果 10
2.3.3 AB堆叠双层蓝磷的电子能带结构和态密度计算结果 11
2.4 本章小结 11
第三章. 单层黑磷烯纳米条带的电子能带结构 12
3.1 紧束缚近似计算方法 12
3.2 黑磷烯纳米条带晶格结构 13
3.2.1 黑磷烯纳米条带的晶格结构 13
3.3 单层黑磷烯纳米条带的电子能带结构 14
3.3.1 具有Zigzag边界的黑磷烯纳米条带电子能带结构 14
3.3.2 具有Armchair边界的黑磷烯纳米条带电子能带结构 15
3.4 本章小结 15
第四章. 总结与展望 16
4.1 总结 16
4.2 展望 16
参考文献 17
致谢 18
磷烯类二维材料的电子能带结构计算
杨斌
20141306026
,China
Abstract:Two dimensional material is born with the discovery of graphene, as a new kind of material has broad application scope and market, we aim to calculate the electron energy band structure and state density of single double-layer blue phosphorus and analyze it by using the first principle method. The electron energy band structure of monolayer black phosphate nano strip is studied by using the method of tight binding approximation. Through the analysis of the electronic energy band structure and the local density graph, we obtain a few layers of blue phosphorus as the indirect bandgap semiconductor. By analyzing the electron energy band structure of the black phosphate nano strip, we found that the sawtooth type black phosphate nano strip shows the metal characteristic, and the armchair type Black phosphate nano strip is an indirect bandgap semiconductor, Very different.
Key words:The first principle, the tight-binding model, the few-layer blue phosphorus, the monolayer black phosphate nano strip, the electron energy band structure, the density of the state.
绪论
- 二维材料介绍
材料在我们日常的生产生活中扮演着非常重要的角色,其推动着科学技术不断的发展。2004年,两名俄裔英籍物理学家英国曼彻斯特大学的安德烈·盖姆和康斯坦丁·诺沃肖洛夫利用机械剥离的方法成功地将这种只有单个原子厚的碳材料即石墨烯,从石墨中剥离出来,两人还因此获得了诺贝尔奖,自此二维材料进入了人们的视野。二维晶体材料因其优异的电子特性,成为半导体材料研究的新的宠儿。
二维材料是指电子仅可在两个维度的非纳米尺度(1-100nm)上自由运动(平面运动)的材料[17]。也就是说,它的厚度被限定在一个原子层厚,但其宽度和长度是无限的。
二维材料拥有许多优越的特性,使它在在能源贮存、吸附、催化、光电等方面展现出巨大应用潜力。
以石墨烯为例,单层石墨烯具有高体征迁移率、大比表面积、优异的力学强度、高透光性等优点。它可以被制成触摸显示屏、发光二极管,通过打开石墨烯的带隙可制成场效应晶体管。
尽管石墨烯具有优良的电学性能,但它没有半导体能隙,基于石墨烯的场效应管也就无法打开跟关闭,这样就失去了场效应管的一个基本功能。在随后发现的MoS2,是直接带隙半导体,也引起人们的研究热潮。但是实验测量发现MoS2的载流子迁移率仅仅可以达到~200cm2/(V*S)。因此,进一步发现具有新型功能并且可实际应用的二维材料是十分有意义的。
蓝磷介绍
元素磷以白色、红色、紫色、蓝色和黑色同素异形体最为稳定, 其颜色由其基本带隙定义。一种具有更宽带隙的稳定同素异形体是蓝磷,其中平面六角结构和大块层叠加非常类似于石墨,相比于黑磷具有较大的带隙,它具有 2 eV 宽的基本带隙。2014年蓝磷被学者预言可稳定存在,并在两年后通过分子束外延的方法在Au(111)衬底上制备出了单层蓝磷,也是一种层状的二维结构。研究者发现的这种磷的同素异形体有着与黑磷相似的热力学稳定性,少数层蓝磷都为间接带隙半导体,其带隙宽度随着层数的增加而降低。这填补了石墨烯没有带隙的遗憾,为二维材料进军半导体材料注入了新的力量,虽然蓝磷作为一种新预测的二维材料才刚刚起步,国内外学者也很少有对蓝磷的研究,但蓝磷优秀的电子特性必然会引起新的研究热潮,这也是我们对这类磷烯类材料进行研究的原因之一。
黑磷介绍
元素磷中最稳定的一个是黑磷, 最近引起了广泛的关注, 因为它在高性能的电子设备的制造中具有很大的潜力。这是因为黑磷具有很高的各向异性,黑磷的带隙是0.59–1.51 eV。
石墨烯因为其自身结构没有带隙,限制了它在半导体产业和光学器件等方面的应用。与石墨烯具有类似的二维层状结构的黑磷,于2014年由中国科学技术大学陈仙辉教授团队使用机械剥离法首次获得。由于其超高的载流子迁移率(~1000cm2/(V*S))和可调控的能隙(直接带隙),它将有可能在未来替代硅,成为电子线路的基本材料。陈教授团队进一步指出,黑磷二维晶体有非常高的漏电流调制率(是石墨烯的10000倍),电流-电压特征曲线展现出良好的电流饱和效应。这些优良的电子性能表明,黑磷相关材料在纳米电子器件应用领域具有极大的潜力。
单层黑磷,又称为黑磷烯,是由单层的磷原子制成的二维晶体。单层黑磷的每一个原胞包含四个磷原子,每个磷原子和其他三个相邻的磷原子组成具有各向异性的褶皱六边形蜂窝状结构。多层黑磷由若干单层黑磷堆叠形成,一般堆叠不超过五层。不同于石墨烯的零能带隙,黑磷是直接带隙半导体,其带隙宽度随着层数的增加而降低,其中单层黑磷的带隙宽度约为1.5eV,五层黑磷的带隙宽度约为0.6eV。相对于蓝磷这样的新面孔,它的老前辈黑磷已经被许多学者所关注并进行研究,所以我们此次只对其单层纳米条带的电子特性进行了解和分析。
本文的工作
本论文是以磷烯类二维材料为研究对象。蓝磷、黑磷是新型的二维材料,与二维材料的开山鼻祖石墨烯不同,磷烯类二维材料多存在带隙,其中蓝磷为间接带隙,黑磷为直接带隙,作为二维材料家族的新成员,关于磷烯类二维材料的研究较之石墨烯、二硫化钼这类大热的二维材料只有很少的一部分,我们此次只对单层蓝磷,AA堆叠双层蓝磷、AB堆叠单层蓝磷、锯齿型黑磷烯纳米条带和扶手椅型黑磷烯纳米条带的部分电子特性进行了分析研究。
研究内容分为以下五个章节在论文中叙述:
第一章为绪论部分,主要介绍本文所研究的磷烯类二维材料的相关信息。
第二章分析少数层蓝磷的能带结构和态密度,包括单层蓝磷和双层蓝磷。
第三章分析黑磷烯纳米条带的能带结构,包括锯齿型黑磷烯纳米条带和扶手椅型黑磷烯纳米条带。
第四章对我们分析所得出的结果进行总结,并对材料未来的发展和研究进行展望。
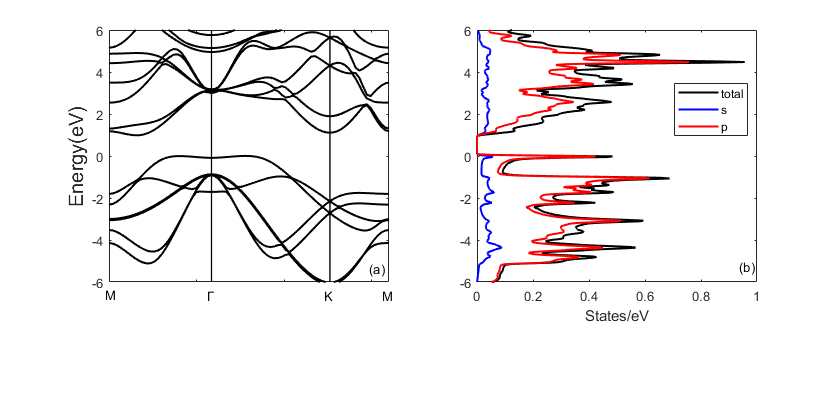
少数层蓝磷的电子能带结构和态密度
- 计算方法
第一性原理计算方法
第一性原理(First Principle)是计算化学或计算物理的一个专业名词,又被称为从头算法[18],指的是所有基于量子力学的计算。利用电子与原子核的相互作用现象,计算分子结构或能量的方式被称作量子计算,通过这种方法我们就可以计算得到物质的各种电子性质。
第一性原理经常是联系计算,在进行计算时,不依靠其他任何经验或半经验参量,也不依靠其他任何实验参量,只给程序所需要的电子或者原子的位置。经验参数是通过大量实例总结出的具有规律性的数据,但是第一性原理却是通过推导演算得出或硬性规定的结论为理论数据。
狭义的第一性原理计算是在计算的过程中不利用经验或半经验的参数,而只参考光速、质子质量或电子质量等实验的数据。但为了加快其计算速度,我们可以添加一些经验参数,当然这样做,第一性原理计算的结果的精度就会不可避免的降低了。
密度泛函理论
密度泛函理论是一种基于量子力学的从头算理论,人们把基于密度泛函理论的计算叫做第一性原理计算[18]。
在化学计算领域和物理计算方面,密度泛函数理论有着十分广泛的应用,是非常有效地研究多电子体系电子结构的量子力学方法。
密度泛函理论可以将多电子的问题转化成单电子的问题,通过基本变量的替换,用电子密度来替换多电子波函数,这样就可以将多电子波函数以及相互对应的薛定谔方程,转化成单电子密度方程。
基于平均场近似的HF方法和超越平均场近似的post-HF方法统称为基于波函数的方法。这类方法通过各种近似方法求解薛定谔方程,得到体系的波函数和能级;再进一步计算体系的各种性质;目前此类方法已经研究极为成熟。
我们知道,近似计算的计算量是于Fock空间中的子空间F(M,N)的维数决定的,而该子空间的维数是随体系尺寸的变化增长的,对于较为精确的计算方法更是这样。计算的复杂程度由波函数的形式来决定。我们假设多电子体系为N电子体系,当N增大的时候,它的的电子波函数具有4N个变量,当然会更加复杂。另一方面,我们知道空间中的势场是可以通过电子密度来算出的,从而我们就可以把体系的哈密顿量和波函数确定了。电子密度就是指电子在空间中的分布函数,变量只有三个,若使用电子密度,而不是波函数,就会将使问题变的简单,并且也能很好的描述体系的状态。H-K就是发现了这一情况,从而为从电子密度出发描述体系性质计算方法奠定了理论基础。
剩余内容已隐藏,请支付后下载全文,论文总字数:14916字
相关图片展示:
该课题毕业论文、开题报告、外文翻译、程序设计、图纸设计等资料可联系客服协助查找;

