论文总字数:12765字
目 录
1.绪论………………………………………………………………4
1.1金属玻璃的概念………………………………………………………………4
1.2金属玻璃的性质与应用………………………………………………………4
1.3金属玻璃的研究现状………………………………………………………………5
2.分子动力学模拟…………………………………………………6
2.1 分子动力学模拟概念…………………………………………………………6
2.2 基本原理………………………………………………………………………6
2.3 分子动力学模拟步骤…………………………………………………………7
3. LAMMPS介绍……………………………………………………9
3.1 LAMMPS简介……………………………………………………………………9
3.2 LAMMPS的功能…………………………………………………………………9
3.3 LAMMPS的优缺点………………………………………………………………9
3.4 lammps的使用…………………………………………………………………10
4.铜锆比例相同的分子动力学模拟………………………………11
4.1in文件分析……………………………………………………………………11
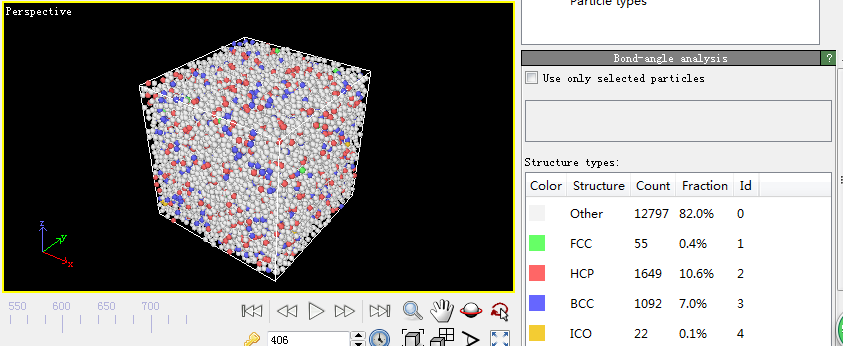
4.2 利用Ovito分析………………………………………………………………12
4.3 RDF分析………………………………………………………………………16
4.4本章小结………………………………………………………………………16
5.铜锆比例不同的分子动力学模拟………………………………17
5.1 RDF分析………………………………………………………………………17
5.2键对分析………………………………………………………………………20
5.3本章小结………………………………………………………………………20
6.结论…………………………………………………………21
参考文献………………………………………………………22
致谢……………………………………………………………23
第一章 绪论
1.1金属玻璃的概念 [1]
从古至今,我们先后经历了石器、青铜、铁器以及现在的新材料时代。我们都知道工具的使用是人类区别于动物的一个重要特征,而工具的不断升级发展对人类社会的发展也起到了巨大的推动作用。要使得工具不断进步,我们就不得不提到新材料的发现与研究。在新材料中,金属玻璃因其独特的结构、优秀的性能引起了世界范围内的广泛关注。
金属玻璃又叫做非晶态合金,它是最近被科学界发现的新材料。金属玻璃的强度要比一般玻璃高得多。这种新型的合金不仅拥有弹性高的特点,而且它的模量还很低。所谓的“金属”,是指这种新型合金材料是由金属原材料通过现代的炼金技术熔炼而成的。而“玻璃”也不是普通意义上我们日常生活中常见的“玻璃”,是指这种新材料的结构是玻璃态式的结构。通常来说,玻璃这种材料在由液态变成固态的过程中不会经过结晶这一过程,而金属合金则会经历这一个过程。我们经常看到的铁制品,金银等贵金属它们的原子排列是有序的,它们属于晶态金属。当我们通过一些技术将高温金属快速降温时,原子会来不及恢复到本来的有序状态留存下来,这个时候原子呈现随机的、无序的排列状态,在微观结构上来看更像是密度非常高的液态,而从宏观上来看又与一般金属一样看不出什么差别,这就是金属玻璃(又叫非晶态合金)。
1.2金属玻璃的性质与应用
金属玻璃具备在长程体现出没有顺序,在短程上体现出有顺序的特点,由此,它具有很多优秀的性能。首先来看合金在力学方面的表现,众所周知,合金在力学方面主要看它的强度和塑性还有导电性能。经过各种实验,我们发现新型大块非晶合金的强度要院大于同类晶态合金。举个例子[3],Mg基非晶合金的抗张强度在室温下高达六百多兆帕斯卡,远远超过抗张强度最大的晶态Mg基合金。而Zr基大块非晶的显微硬度其强度已接近工程陶瓷材料的程度。在塑性方面,我们甚至可以将非晶态合金吹制成一种球状物体,可见它的塑性之强。
在电导方面,由于非晶态合金有着像液体一样均匀的特点,它表现出和晶态合金各向异性完全相反的特性--各向同性,当然它的导电性能就很差。 金属玻璃有还有着低磁损耗的特性,因为这个特性,它可以在变压器中代替硅钢片作为铁芯。这样能够减少高达三分之二的电能损耗。
由于金属玻璃还拥有耐磨,抗辐射,耐腐蚀,低热导,高声阻抗等特点。它在军事、航天方面也发挥着重要的作用。比如说制作复合装甲来装备坦克,在宇宙飞船上使用金属玻璃来收集太空中的特殊物质以研究我们所处的神秘未知的宇宙。
1.3金属玻璃的研究现状[5]
非晶态合金具有如此多的优秀性能,吸引了一大批的国内外优秀科学家争相研究它以期发现更多具有高使用价值的新材料。正因如此,非晶态合金的研究这些年也取得了巨大的进展。中国科学院物理研究所经过不懈的努力,花了十几年的时间努力钻研,发现了性能优秀的金属塑料;在根据弹性模量判据探索新型稀土非晶材料方面也取得了一些优秀的成果。2011年1月14日,在国家科学技术奖励大会上,中国科学院物理研究所非晶材料和物理研究组独立完成的“非晶合金形成机理研究及新型稀土基块体非晶合金研制”项目荣获2010年度国家自然科学二等奖。中国科学院物理研究所非晶材料和物理研究组一直在从事高性能金属玻璃的设计、内部结构及性能表征方面的研究工作。通过近十几年的研究,该组发展了通过调控非晶材料弹性模量来控制非晶合金性能和形成的弹性模量判据;发现了兼有金属和塑料重要特性的非晶合金材料—— 金属塑料。
虽然现在对金属玻璃的研究已经广泛开展起来,但是和已经发展成熟的晶态金属比起来还处在起步阶段,仍然存在着许多问题需要我们去探索解决。就目前而言,非晶态研究方面还存在一些4个方面的不足[4]:
1、非晶形成能力及非晶合金的成分设计。
2、非晶合金的结构及结构模型。
3、非晶态合金的形变机理。
4、脆性断裂问题。
第二章 分子动力学模拟
2.1 分子动力学模拟概念[2]
分子动力学模拟(Molecular Dynamics (MD) Simulation)的基本思想是通过粒子微观体系的特性计算来反映体系的宏观特性。它的核心方法是在一个有边界的范围内设立一些粒子,并将它们看成质点,通过牛顿运动方程来计算出这些质点在不同时刻的坐标以及速度,以此来观察体系内粒子的状态。它是一种计算机模拟研究方法。[12]这种方法由安德尔和威尔怀特在1957年提出,发展至今先后增加了Verlet算法,Predictor correctoer算法,Anderson定压法,damp force法,第一性原理分子动力学方法等。涵盖的算法越来越多,成为科学家们研究新材料时的一个重要选择方法。它被广泛应用于高温液态物体迅速降温过程中对粒子所处状态的研究,很符合本文所作内容的需要。
2.2 基本原理[6]
首先选取N个原子,得到它们T时刻所处的状态(位置,速度等参数)。我们可以通过粒子间的相互作用力得到,每个原子此时受到的力的大小。选取一个极其微小的间隔△t,通过F=ma得到每个原子经过的位移,来得到t △t时刻系统的状态,当△t趋向于0时我们就越接近一个准确的结果,不断重复这一个过程,我们就可以得到系统随时间的演变过程,而分子动力学中的位移方程可以通过下方经典的Lagrange方程得到[8]
(2.1)

上式中,mi和ri分别为第i个粒子的质量和位矢,V(rij)为作用势。一般情况下,方程在确定的时间步长内积分求解,从而得到下一时间步长粒子的位置,因此,在初始构型及确定的边界条件下,分子动力学的计算结果反映了在一定时间间隔内粒子在经典力学框架下的运动轨迹。从而可以获得体系的统计力学以及动力学方面的信息。
2.3 分子动力学模拟步骤
动力学的步骤实际上可以分为4步[7]:
1.先要设定一个模拟要采用的模型以及势函数。
2.给定初始条件。
3.趋于平衡的计算过程。
4.宏观物理量的计算。
第一步,我们要选取一个合适的时间步长△t。△t的选取可能会影响模拟的结果甚至会导致实验误差过大或者失败,也可能会让模拟的效率过低。所以△t的选取要参考分子的特征运动频率。虽然对于它的选取没有一个明确的标杆,但是我们需要遵照一个准则:总的能量波动高于势能波的几个百分点。
然后我们确定要用到的势函数。由于势函数的形式有许多种,不同的势函数作用的对象又不同,所以势函数的选定在分子动力模拟中至关重要。
第二步,给定初始条件。在一定温度T的平衡系统中,粒子的初始速度一般采用麦克斯韦[16]分布形式(2.2)

其中l表示速率的分量,然后会产生随机数vl来满足上式,将这个随机数给予各个粒子,我们就得到了粒子的起始速度。
第三步,趋向平衡的计算过程。得到了粒子的开始速度以后,在这个基础上再看粒子的受力情况,调整粒子的相空间坐标使其慢慢到达平衡状态。这个平衡过程在分子动力学模拟中叫做弛豫,这是一个很必要的过程。在进行弛豫的过程中,体系中的原子间又会产生相互作用力,有了这个相互作用力,体系最终趋于平衡。同时原子之间的作用也决定了弛豫的过程的宏观规律。弛豫和系统的无序是紧密相连的,因为它展示的是一个消耗能量的过程。
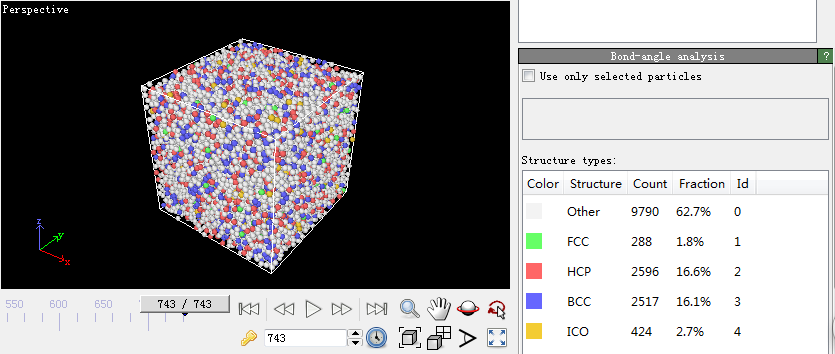
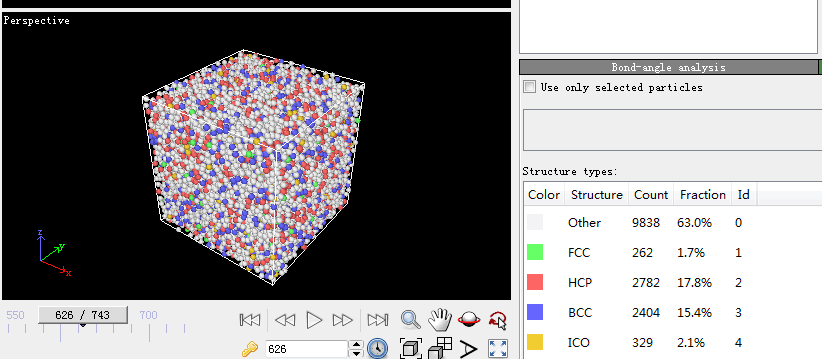
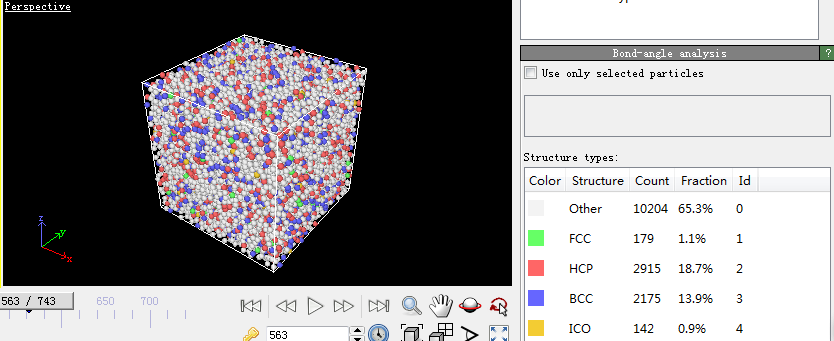
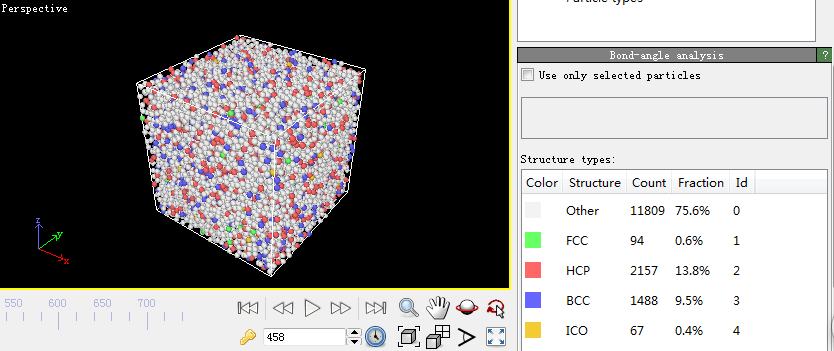
第四步,宏观物理量的计算。因为在模拟的过程中只能看到一些粒子所处位置以及它们的速度随着温度和时间发生变化,看不出它们所反映出来的规律,因此一些能反映这些规律的观察方法被提了出来。比较常见的有均方位移(MSD)、径向分布函数(RDF)、系统总能量、H-A键型指数分析技术和VMD分子动力学可视化方法。本文用到的径向分布函数(RDF)。下面是关于它的介绍:
RDF是表征金属玻璃结构的最重要的函数。它与X射线衍射实验得到的干涉函数互为傅立叶变换。它的物理意义:以一个粒子为中心,在半径为r到r dr的空间范围里发现另一个粒子的概率。通常定义为:
(2.3)

用以说明结构的无序化程度。
其中:p代表系统的平均密度,R表示的是原子所处坐标,括号中的公式表示的是time平均,δ代表Dirac函数,N表示原子数量。RDF函数与结构因子互为傅立叶变换:

(2.4)

径向分布函数是理论与实验对照的基本依据。
第三章 LAMMPS[13]介绍
3.1 LAMMPS简介
LAMMPS是由美国一家实验室开发的开源软件,它是一个经典的分子动力学代码,可以进行绝大多数的 分子动力学模拟。LAMMPS可以计算的粒子数可以很少也可以达到以亿为单位的数量。它可以用来模拟液体中的原子状态,也可以模拟固、气体的系综。它是专门为并行计算机设计的,虽说如此,但它其实也可以在硬件不是很好的台式机或者笔记本上以不错的计算效率运行。由于它是一个开源软件,它的源代码可以被使用者根据自己的需求来改动。LAMMPS具有很好的并行扩展性。
3.2 LAMMPS的功能[15]
剩余内容已隐藏,请支付后下载全文,论文总字数:12765字
相关图片展示:
该课题毕业论文、开题报告、外文翻译、程序设计、图纸设计等资料可联系客服协助查找;

